6729742
Description
Mind Map by Xareni Rebollar Juárez, updated more than 1 year ago
|
|
Created by Xareni Rebollar Juárez
almost 8 years ago
|
|
NEFROPATIA
POLIQUISTICA AR
(INFANCIA)
- DEFINICION:
- Trastorno multisistémico en el que
aparecen y se desarrollan quistes en
el tejido renal, que progresivamente
ocupan el parénquima normal junto
con zo- nas de fibrosis e inflamación
intersticial.
- Trastorno multisistémico en el que
aparecen y se desarrollan quistes en
el tejido renal, que progresivamente
ocupan el parénquima normal junto
con zo- nas de fibrosis e inflamación
intersticial.
- DEMOGRAFIA:
- Afecta a
neonatos
- Afecta a
neonatos
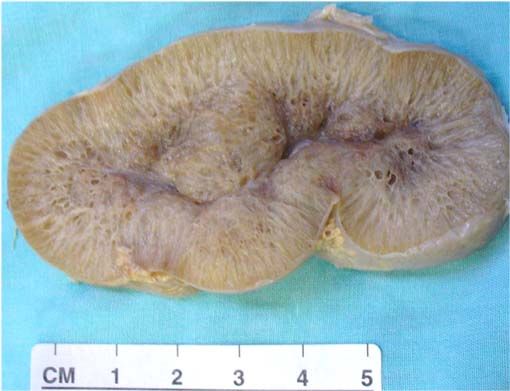
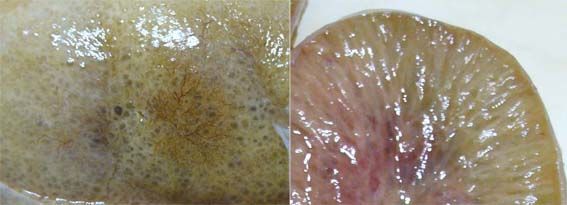
- MORFOLOGIA MACROSCOPIA:
- Riñones están aumentados de tamaño y
tienen aspecto externo normal. Al corte
quistes pequeños en la corteza y la medula
dan aspecto en esponja. Canales alargados y
dilatados forman un ángulo recto con la
superficie cortical, reemplazando medula y
corteza
- Apariencia
espongiforme
generalizada, el
aspecto radiado de
las dilataciones
quísticas y la sutil
diferencia entre la
corteza y la médula
- Apariencia
espongiforme
generalizada, el
aspecto radiado de
las dilataciones
quísticas y la sutil
diferencia entre la
corteza y la médula
- Riñones están aumentados de tamaño y
tienen aspecto externo normal. Al corte
quistes pequeños en la corteza y la medula
dan aspecto en esponja. Canales alargados y
dilatados forman un ángulo recto con la
superficie cortical, reemplazando medula y
corteza
- PRONOSTICO
- La supervivencia inicial depende del grado de hipoplasia pulmonar. El
requerimiento de ventilación mecánica se correlaciona con la
mortalidad. Sólo 3 de 49 pacientes seguidos una media de 8,46 ± 6,24
años fallecieron después del primer mes de vida. A más largo plazo,las
tasas de supervivencia a los 15 años oscilan entre el 50 y el 80%
- La supervivencia inicial depende del grado de hipoplasia pulmonar. El
requerimiento de ventilación mecánica se correlaciona con la
mortalidad. Sólo 3 de 49 pacientes seguidos una media de 8,46 ± 6,24
años fallecieron después del primer mes de vida. A más largo plazo,las
tasas de supervivencia a los 15 años oscilan entre el 50 y el 80%
- GENETICA
- Está implicado un gen localizado en
el brazo corto del cromosoma
6p21.1-p12, llamado PKHD1. Es
autosómica recesiva. La mayoría
son heterocigotos compuestos.
- Está implicado un gen localizado en
el brazo corto del cromosoma
6p21.1-p12, llamado PKHD1. Es
autosómica recesiva. La mayoría
son heterocigotos compuestos.
- IMPACTO:
- Tiene una incidencia de
1:20,000 recién nacidos vivos y
se manifiesta produciendo una
patología severa durante la
gestación o en los primeros
meses de vida.
- Tiene una incidencia de
1:20,000 recién nacidos vivos y
se manifiesta produciendo una
patología severa durante la
gestación o en los primeros
meses de vida.
- TRATAMIENTO
- Sintomático y de soporte. En periodo neonatal es frecuente realizar
maniobras de resucitación y de soporte ventilatorio. Casos que
asocian hipoplasia pulmonar pueden ser incompatibles con la vida. La
HA es frecuente y de aparición en fases tempranas . Responde bien a
IECAS y bloqueadores de los canales del Ca. La IR se trata de manera
conservadora, previniendo en la manera de lo posible la
osteodistrofia renal, tratando la anemia y el retraso de crecimiento.
El tratamiento definitivo es el TRANSPLANTE RENAL,
- Sintomático y de soporte. En periodo neonatal es frecuente realizar
maniobras de resucitación y de soporte ventilatorio. Casos que
asocian hipoplasia pulmonar pueden ser incompatibles con la vida. La
HA es frecuente y de aparición en fases tempranas . Responde bien a
IECAS y bloqueadores de los canales del Ca. La IR se trata de manera
conservadora, previniendo en la manera de lo posible la
osteodistrofia renal, tratando la anemia y el retraso de crecimiento.
El tratamiento definitivo es el TRANSPLANTE RENAL,
- ETIOLOGIA:
- Mutación del gen PKHD1
- Mutación del gen PKHD1
- FISIOPATOLOGIA:
- El gen PKHD1 codifica la
fibrocistina, proteína integral
de la membrana, posible
receptor de superficie celular
que participa en diferenciación
del túbulo colector y biliar
- El gen PKHD1 codifica la
fibrocistina, proteína integral
de la membrana, posible
receptor de superficie celular
que participa en diferenciación
del túbulo colector y biliar
- CLINICA
- Desarrollan una peculiar lesión hepática que
se caracteriza por fibrosis periportal blanda y
proliferación de conductillos biliares bien
diferenciados: fibrosis hepática congénita.
Tienen hipertensión portal con
esplenomegalia, varices esofágicas, retraso
en el crecimiento, dificultad para
alimentación, infección urinaria.
- Desarrollan una peculiar lesión hepática que
se caracteriza por fibrosis periportal blanda y
proliferación de conductillos biliares bien
diferenciados: fibrosis hepática congénita.
Tienen hipertensión portal con
esplenomegalia, varices esofágicas, retraso
en el crecimiento, dificultad para
alimentación, infección urinaria.
- AUXILIARES DIAGNOSTICOS:
- Hallazgos ecográficos más característicos son aumento del
tamaño renal, oligoamnios y falta de llenado vesical,
también nefromegalia bilateral, múltiples pequeños quistes,
- Hallazgos ecográficos más característicos son aumento del
tamaño renal, oligoamnios y falta de llenado vesical,
también nefromegalia bilateral, múltiples pequeños quistes,
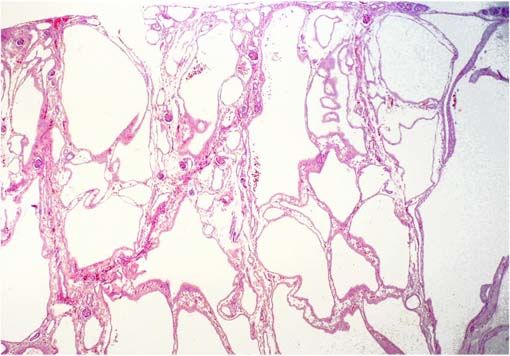
- MORFOLOGIA MICROSCOPICA:
- Dilataciones cilíndricas o saculares de
todos los conductos colectores.
Quistes tienen revestimiento
uniforme de células cubicas
- Múltiples quietes de tamño variavle, el epitelio es
cilíndrico bajo, cúbico o puede verse plano por
compresión. No hay quistes glomerulares. Entre
los quístes hay parénquima renal que puede
mostrar, en algunos casos, fibrosis
- Múltiples quietes de tamño variavle, el epitelio es
cilíndrico bajo, cúbico o puede verse plano por
compresión. No hay quistes glomerulares. Entre
los quístes hay parénquima renal que puede
mostrar, en algunos casos, fibrosis
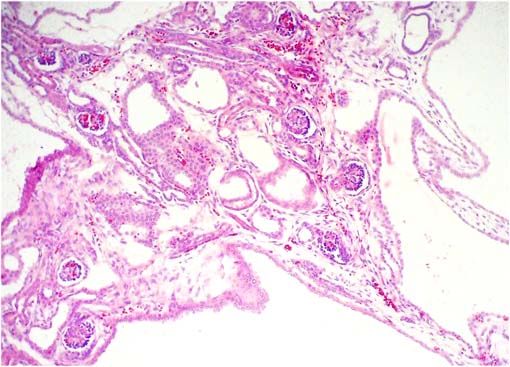
- Cavidades quísticas y túbulos
dilatados, el epitelio puede
verse hiperplásico. Note los
glomérulos preservados, en este
caso con aspecto fetal (el caso
corresponde al de un
mortinato), pero sin dilatación
del espacio de Bowman
- Cavidades quísticas y túbulos
dilatados, el epitelio puede
verse hiperplásico. Note los
glomérulos preservados, en este
caso con aspecto fetal (el caso
corresponde al de un
mortinato), pero sin dilatación
del espacio de Bowman
- Dilataciones cilíndricas o saculares de
todos los conductos colectores.
Quistes tienen revestimiento
uniforme de células cubicas
- METODOS ESPECIALES:
- La genética molecular permite un
diagnóstico fiable a partir de las
semanas 12 de gestación, en el
aproximadamente 80% de la
población de familiares afectos
- La genética molecular permite un
diagnóstico fiable a partir de las
semanas 12 de gestación, en el
aproximadamente 80% de la
población de familiares afectos
- VARIANTES:
- Perinatal,,
neonatal, del
lactante y juvenil
- Perinatal,,
neonatal, del
lactante y juvenil
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.