3911460
Síndrome de von Hippler-Lindau
- ¿Qué es?
- Es una rara entidad genética que se caracteriza por la predisposición
al cáncer, especialmente angiomas de la retina, hemangioblastomas
del sistema nervioso central y carcinoma renal de células claras.
- Se presenta en 1 por cada 35,000 individuos
- Al parecer se transmite como un rasgo
mendeliano autonómico dominante
- Típicamente, los síntomas aparecen de la
segunda a la cuarta décadas de la vida
- Genética
- Por estudios de ligamiento se identificó la banda 3p25 como el "locus"del gen VHL
- La enfermedad, como casi todos los cánceres
hereditarios, se produce por una mutación de
línea germinal de un gen supresor tumoral
(VHL),, es necesaria una mutación somática
posterior para la aparición de los síntomas
- La enfermedad, como casi todos los cánceres
hereditarios, se produce por una mutación de
línea germinal de un gen supresor tumoral
(VHL),, es necesaria una mutación somática
posterior para la aparición de los síntomas
- Las manifestaciones fundamentales para el diagnóstico
clínico de la enfermedad de von Hippel-Lindau (VHL) son:
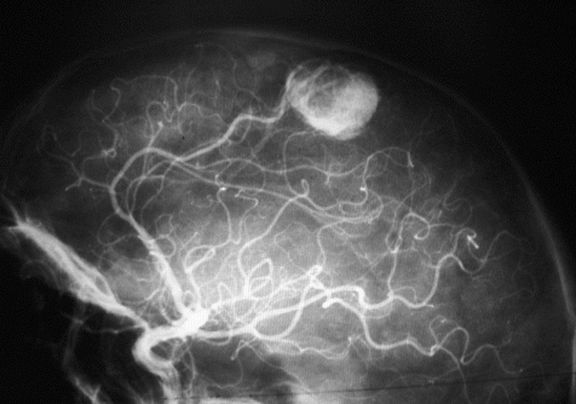
- Angiomas de retina
- Hemangioblastomas del encéfalo
- El examen microscópico de los hemangioblastomas
muestran una mezcla de "células del estroma" y
vasos sanguíneos (pericitos y células enodteliales) y
se ha demostrado que de ellas las células tumorales
son las del estroma
- El examen microscópico de los hemangioblastomas
muestran una mezcla de "células del estroma" y
vasos sanguíneos (pericitos y células enodteliales) y
se ha demostrado que de ellas las células tumorales
son las del estroma
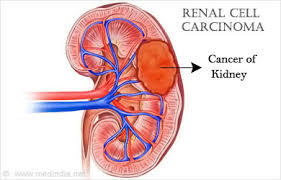
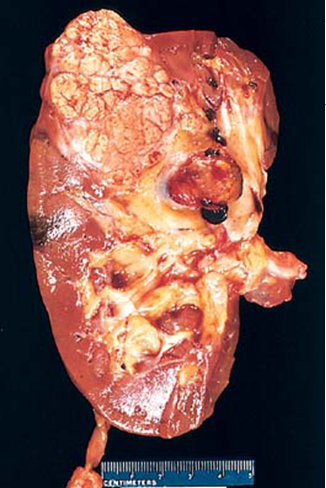
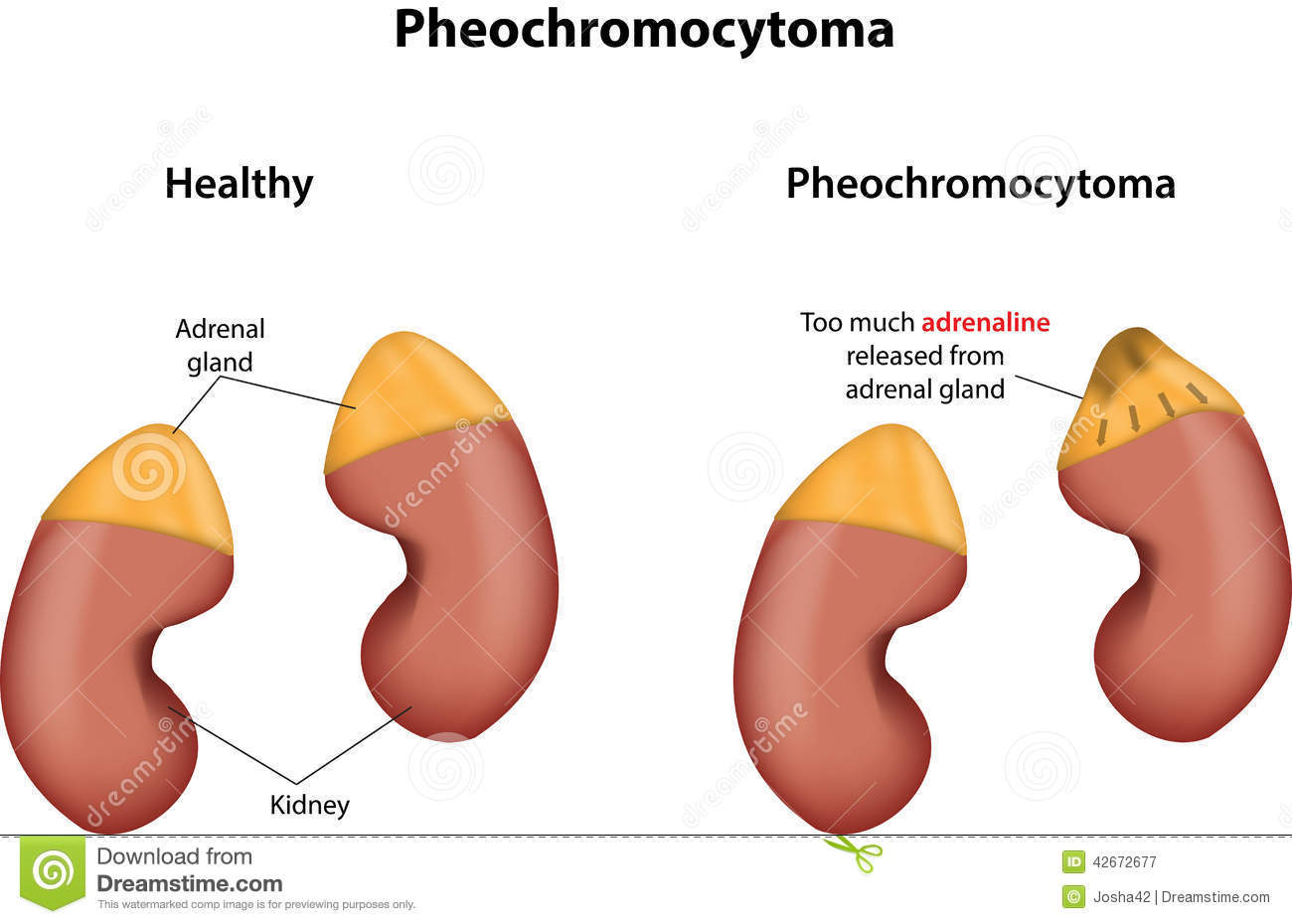
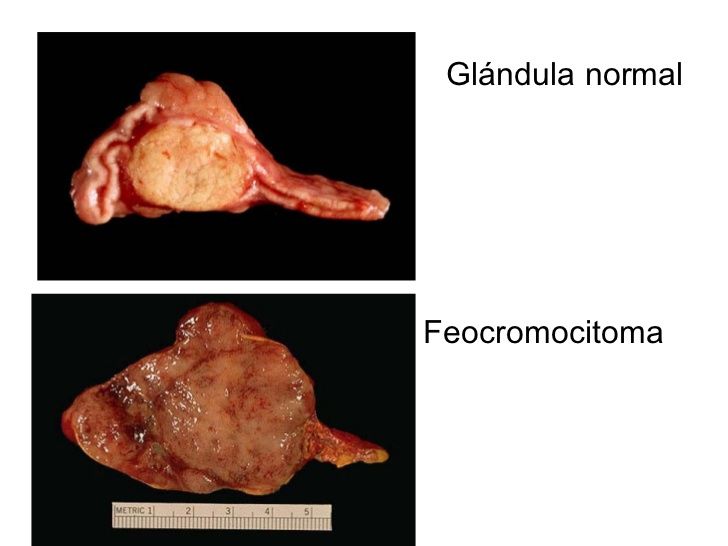
- También son frecuentes los carcinomas de
células renales y menos los feocromocitomas
- Carcinoma renal
- Feocromocitomas
- Carcinoma renal
- Tratamiento
- Los hemangioblastomas retinianos
son tratados con fotocoagulación
con LASER, pero esto puede producir
desprendimiento de la retina y
ceguera
- Se ha empleado la extirpación quirúrgica de los tumores
encefálicos y espinales, pero frecuentemente durante la
operación se han producido daños en la zona que han
llevado a la parálisis del paciente
- La extirpación de tumores renales ha conducido a la insuficiencia
renal. La mayor parte de los pacientes mueren después de los
cincuenta años, principalmente por los hemangioblastomas y los
carcinomas renales
- Los hemangioblastomas retinianos
son tratados con fotocoagulación
con LASER, pero esto puede producir
desprendimiento de la retina y
ceguera
- Angiomas de retina
- Se presenta en 1 por cada 35,000 individuos
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.